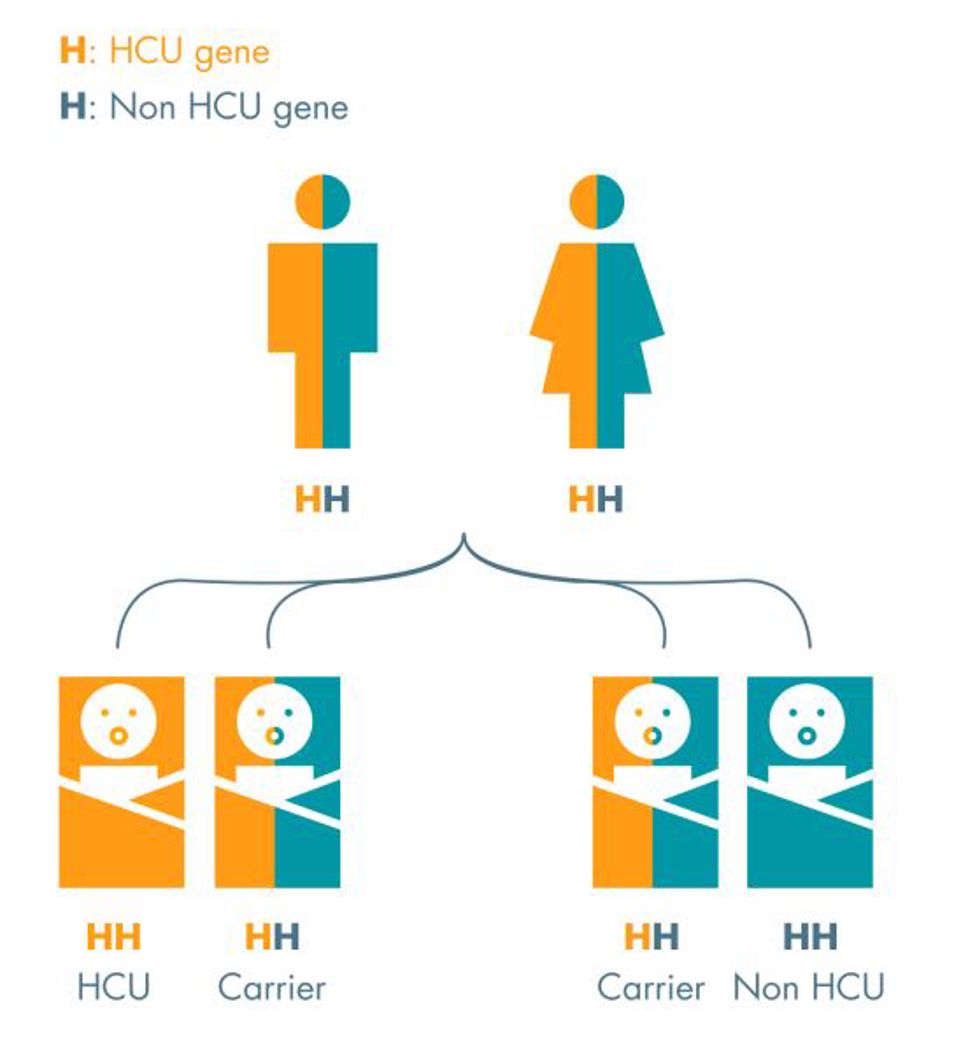
It is variations in the CBS gene that cause HCU. This gene provides the specific instructions for the body to make an enzyme called cystathionine beta-synthase, responsible for the process that breaks down the amino acids homocysteine and methionine into a molecule called cystathionine. Without the enzyme, homocysteine and methionine build up in the blood. This type of HCU is termed “Classic Homocystinuria”.
Occasionally, HCU can be caused by variations in several other genes, the enzymes made by these genes also play a part in the conversion of homocysteine to methionine. Variations in these genes can interfere with the function of the enzymes, also causing a buildup of homocysteine. Experts are yet to determine how excess homocysteine and related compounds lead to the signs and symptoms of homocystinuria.5
Another form of HCU is “Cobalamin (cbl) cofactor metabolism defect”. Normally the body will convert some homocysteine back into methionine, a process that requires changing the form of cobalamin (vitamin b12). Homocystinuria caused by cbl defect occurs when your body is unable to process these changes due to an inability to create the required enzymes effectively.2
Symptoms and diagnosis
Babies born with HCU do not tend to display symptoms for their first year, but without treatment, the following symptoms can develop later in life:
- Thrombosis (blood clots) anywhere in the body
- Dislocation of the lens of the eye
- Severe short-sightedness (myopia)
- Tall stature, long arms and legs
- Brittle & fragile bones (osteoporosis)
- Curving of the spine (scoliosis) & other skeletal abnormalities
- Delayed development and learning difficulties6
Diagnosis is usually quite simple, standard blood spot screening tests at 5 days post-birth on infants will reveal if they have been born with HCU. If detected, treatment can be provided to reduce the risk of serious complications and, if caught early and treated the majority of children with HCU are able to live normal, healthy lives.4
Living with HCU & treatment
Many of the symptoms of HCU are treatable and can be prevented by early and aggressive therapy. For patients with classic HCU, it is possible to control their homocysteine levels using vitamin B6 (pyridoxine). Response to this medication splits classic HCU into 3 classifications:
- Vitamin B6-responsive - body creates enough CBS enzyme, therefore vitamin B6 can help the enzyme operate effectively.
- Partially vitamin B6-responsive - body makes some CBS enzyme, as such vitamin B6 can partially help it operate effectively.
- Vitamin B6 non-responsive - body does not make enough CBS enzyme, so vitamin B6 has no effect.2
If one of the first two classifications, then vitamin B6 supplements must be taken daily for the rest of their lives. Additionally, for patients who have partial responsiveness or no responsiveness to vitamin B6, there are further treatment options, such as medication called betaine (cystadane), special diet, and additional supplements.
The aim of treatments is to keep the body’s level of homocysteine below 100 mcmol/L at all times of day as this can prevent further complications relating to HCU. This goal is even lower for vitamin B6-responsive individuals with a target of 50 mcmol/L, which is more easily achievable without needing to adversely affect nutritional intake.6
How ZOIA Pharma can help
As a Pentec company, ZOIA Pharma is dedicated to serving patients with inherited metabolic disorders and other rare diseases, including those affected by HCU. Alongside vitamin B6 treatment, diet regulation to ensure low levels of protein are eaten is crucial in managing HCU.
Low protein foods naturally contain low levels of protein or they can be specially formulated and processed to remove protein.
ZOIA provides low protein foods as well as other vital nutritional products, such as metabolic formulas, from trusted specialist manufacturers including eTon Pharmaceuticals, Firstplay Dietary Foods (Promin), Vitaflo, Abbott, and Nutricia.
Food is distributed from ZOIA’s 11,000 square-foot U.S. Food and Drug Administration-registered medical food warehouse and storage facility. ZOIA helps keep treatment stress-free for patients and providers by investigating insurance benefits and potential reimbursement, identifying affordable options, providing home delivery, and more. We are able to directly support the rare disease community in a way no other national provider can.
Enjoy seamless health management with clinical support, insurance navigation, and nationwide delivery. That’s the ZOIA difference.
“It has been a struggle for years to get my insurance to pay for my son's medical formula. Your company has made my son's life better as he never has to worry about his medical formula again and your billing team makes the insurance reimbursement process a breeze.”
— ZOIA Parent of an HCU Patient
HCU is a rare condition, but that doesn’t make the treatment or patients any less important. Patients deserve a reliable provider that is able to meet their needs, and that’s why we have remained committed to the rare diseases community with a dedicated team providing a combined 20 years of experience. With ZOIA, HCU doesn’t have to define your story, it’s simply a footnote in a normal, fulfilling life.